Article
Comprehensive analyses of 1771 transcriptomes cross 7 tissues enhance genetic and biological interpretations for complex traits in maize
https://www.biorxiv.org/content/10.1101/2023.08.09.552713v1
Abstract
By analyzing 1771 RNA-seq data from 7 tissues sampled from 298 genotypes, we studied the tissue expression specificity and built a comprehensive multi-tissue gene regulation atlas in maize. We describe the landscape of the transcriptome variation and identify thousands of alleles associations with gene expression in 7 tissues. The tissue-sharing patterns of these genetic regulatory effects is consistence with phenotypic correlation, highlighted a general contribution from tissue specific regulatory variation to across tissue transcriptomes variation. Using transcriptome-wide association study (TWAS), we linked gene expression variation in different tissues to agronomic traits variation, revealing tissue-specific expression contribution to traits. In addition, through integrative analyses of tissue-specific gene regulation variation with genome-wide association studies, we detected relevant tissue types and candidate genes for agronomic traits to elucidate the genetic mechanisms underpinning such agronomic traits in maize. Our findings provided novel insights into the genetic and biological mechanisms underlying complex traits in maize, and our transcriptome atlas can serve as a primary source for biological interpretation, functional validation, and genomic improvement in plants.
Expression diversity across 7 tissues in maize diversity panel
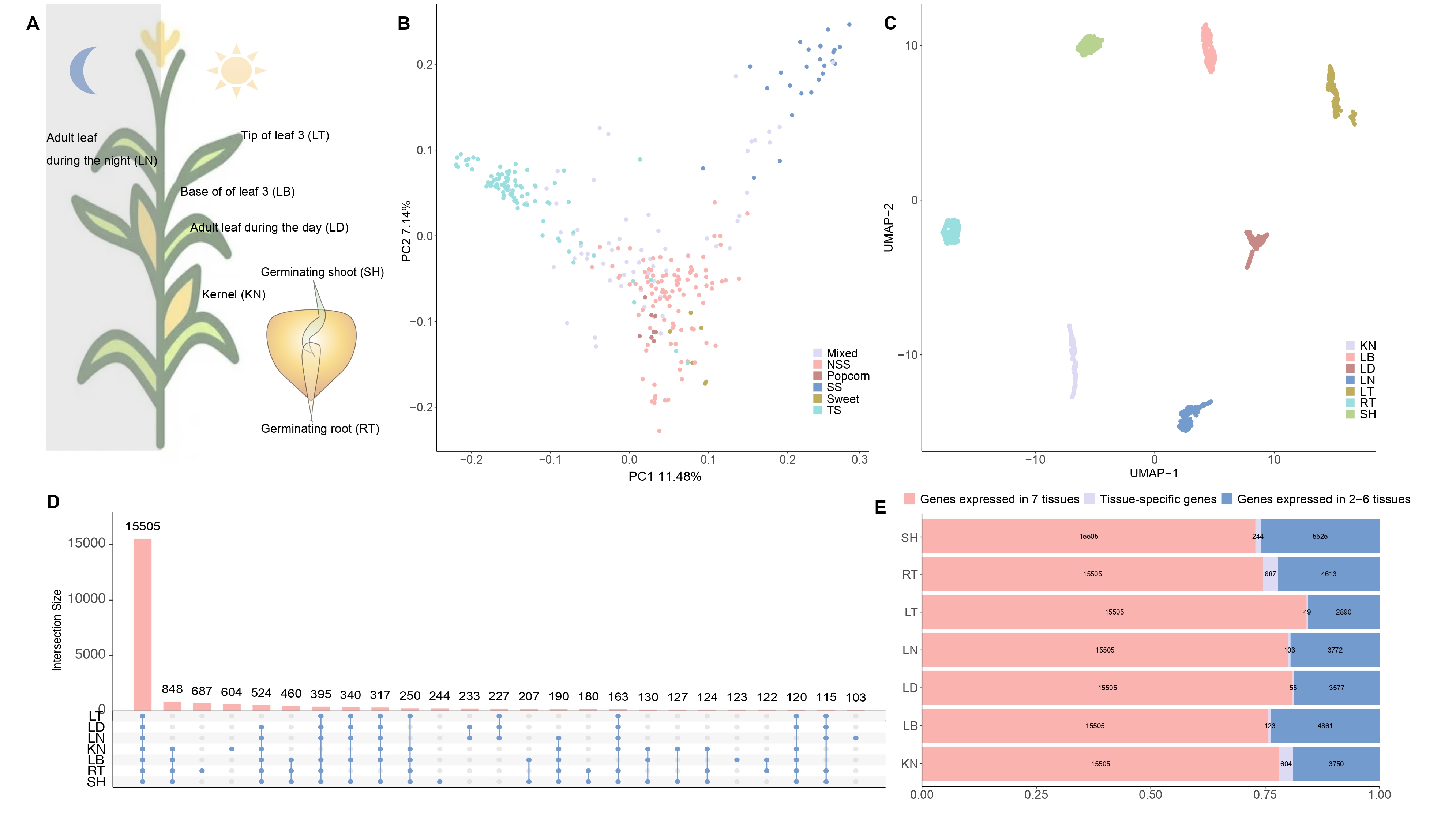
Figure 1. Characteristics of 298 maize samples and expression diversity of 7 tissues. (A) 7 tissues were analyzed. (B) Principal component analysis of 4,260,521SNPs based samples from 6 groups. (C) The UMAP of 7 tissues based on the expression of 15505 genes across samples. (D) The genes that are expressed in 7 tissues overlap. (E) The gene type distribution in each tissue.
Tissue- specific genetic regulation of expression diversity
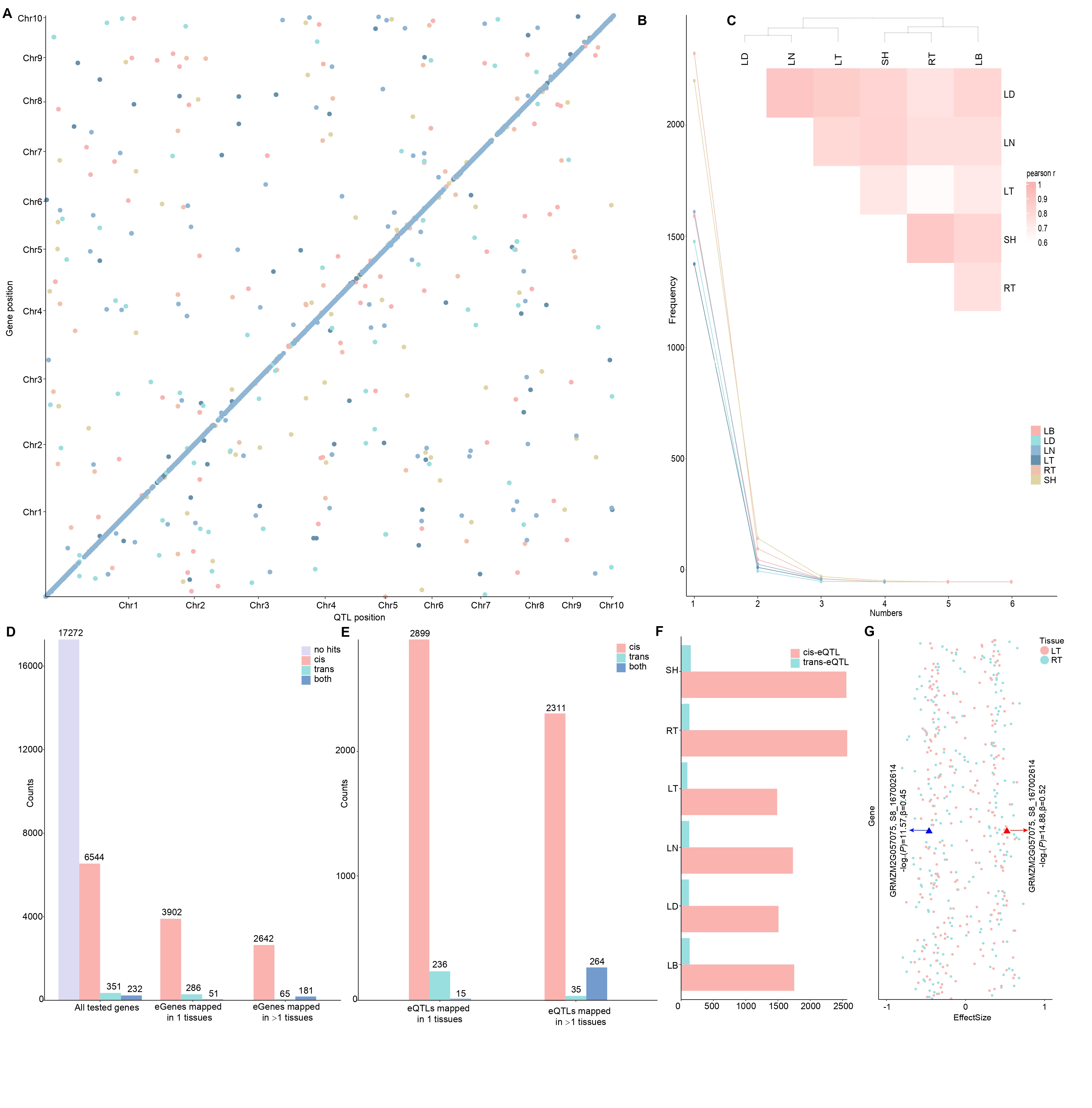
Figure 2. Summary of the eGWAS results. (A) A genome-wide map of eQTLs in 6 tissues. The x-axis shows the location of the eQTLs, while the y-axis shows the position of the associated gene. Points along the diagonal are cis-eQTLs (those mapping to nearby genes). (B) The number of eQTLs associated with one gene in each of 6 tissues. (C) Tissue clustering with pairwise Pearson correlation of eQTL effect sizes. Tissues are grouped by hierarchical clustering. (D) The number of eGenes. (Left) The number of genes with different kinds of eQTL among all genes tested. (Middle) Of the 4239 eGenes mapped in 1 tissue, 3902 were found to be associated with cis-eQTL, indicating a highly significant enrichment. Right Hits that were mapped in multiple tissues were more likely to associate cis-eQTL. (E) The number of eQTLs. (F) The number of cis and trans eQTLs that have been identified in each tissue. (G) The eQTL effects of similar eGene in RT and LT. Triangle symbols represent the top eQTL of GRMZM2G057075.
Tissue expression diversity contributes to trait diversity
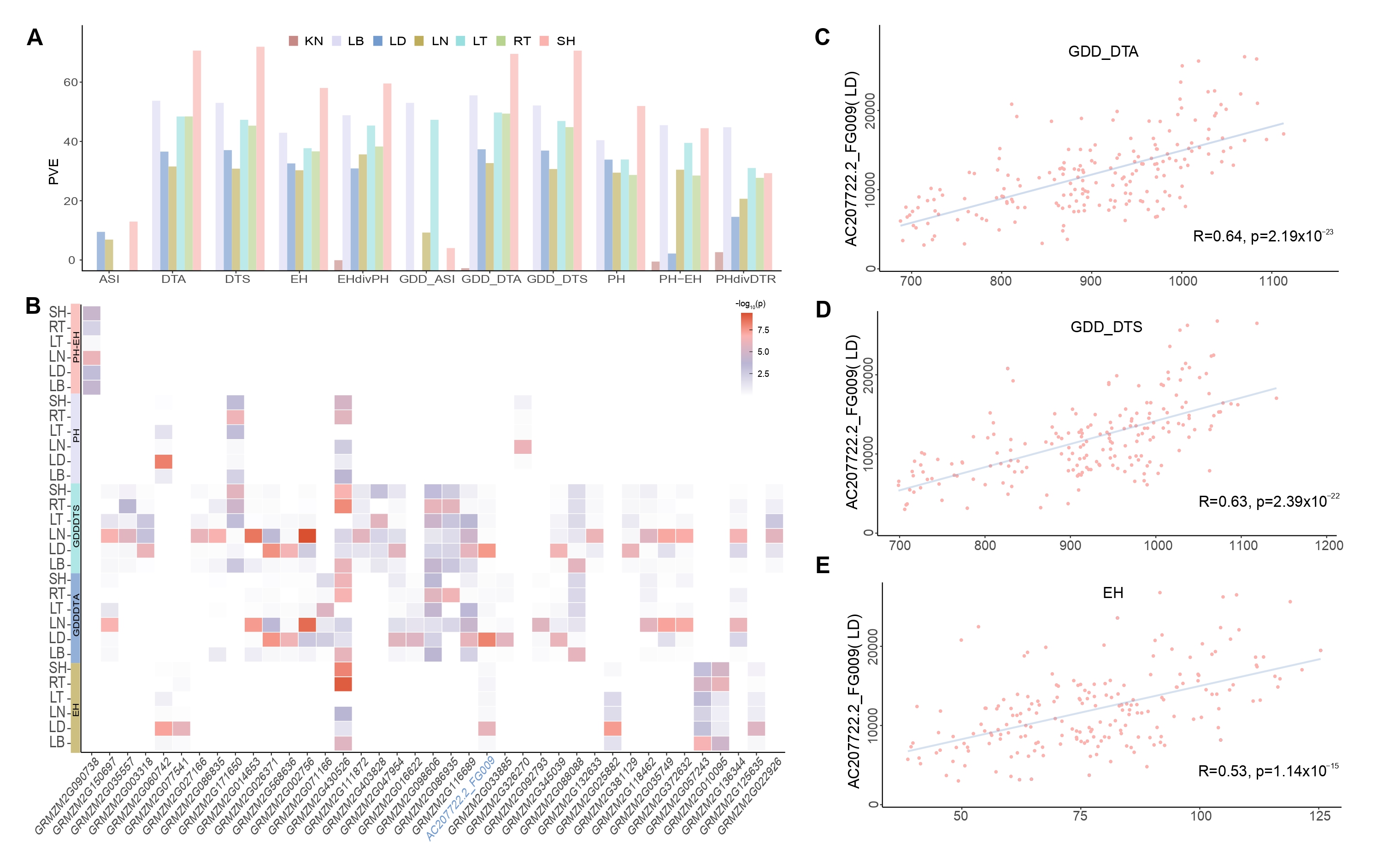
Figure 3. Tissue diversity contributes to trait diversity. (A) The phenotypic variance explained (PVE) by gene expression in 7 tissues (B) Gene associated with agronomic traits in 6 tissues. Each square’s color represents the gene’s P value. (C) and (E) Expression-to-trait correlation maps for AC207722.2_FG009, highlighting gene expression highly positive correlation with GDD_DTA, GDD_DTS and, EH in LD.
Tissue-specific eQTL effects on agronomic traits
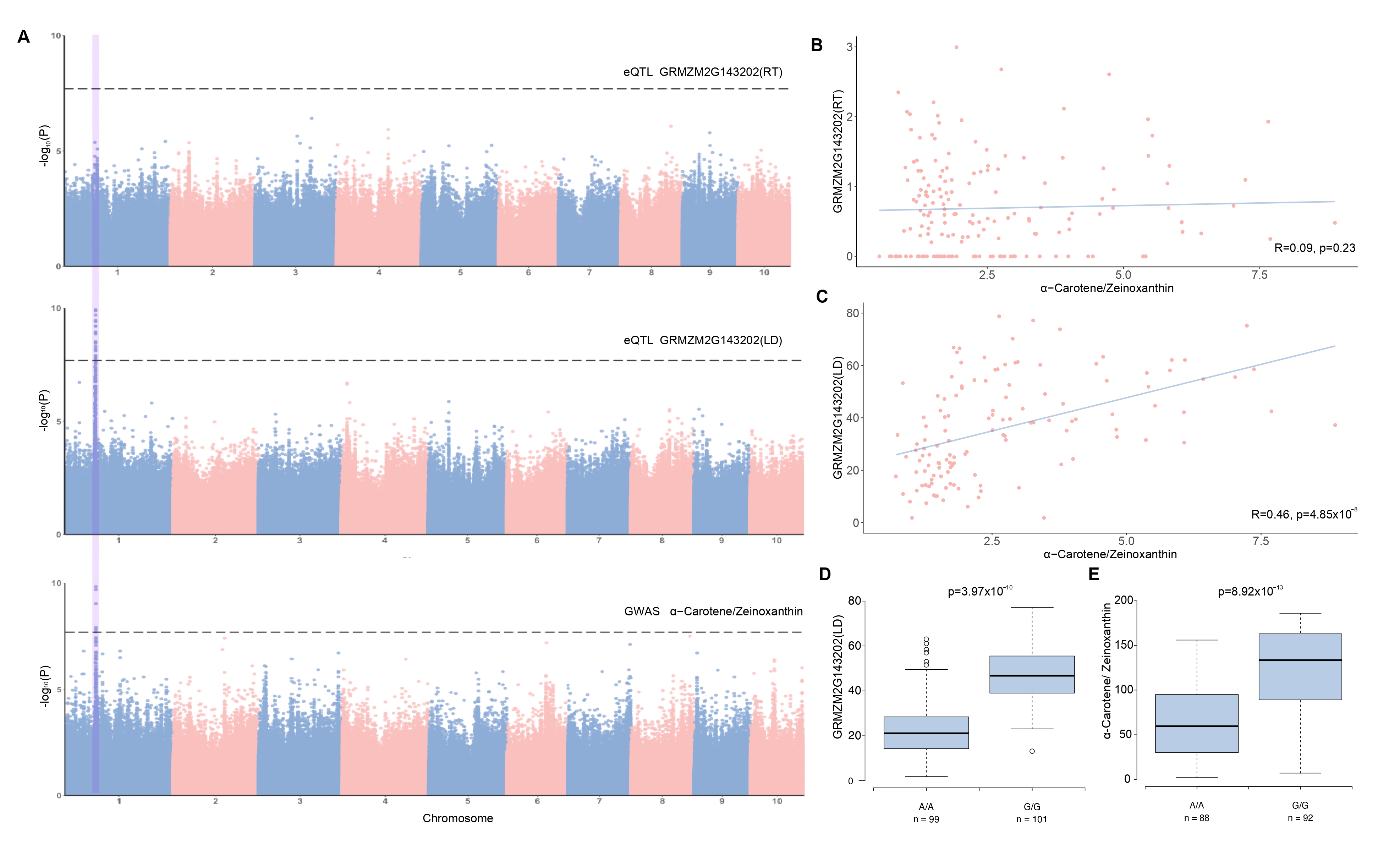
Figure 4. Summary of QTL S1_86775549 associated with α-Carotene/Zeinoxanthin and GRMZM2G143202. (A) Manhattan plots from top to bottom are GRMZM2G143202 in RT, GRMZM2G143202 in LD, and α-Carotene/Zeinoxanthin. The position of S1_86775549 was highlighted, illustrating a tissue-specific association. (B) and (C) Gene expression-to-phenotype maps. GRMZM2G143202 expression in LD was highly positively correlated with α-Carotene/Zeinoxanthin, whereas gene expression in RT was not significantly correlated with the trait. (D) and(E) Genotype-to-phenotype maps. The phenotype (GRMZM2G143202 and α-Carotene/Zeinoxanthin) comparison between two alleles of S1_86775549. P-values were obtained by GWAS.
Identification of the potential tissue-specific regulation mechanism
Table 1 List of the tissues-specific candidate genes associated with agronomic traits and reported biological function.
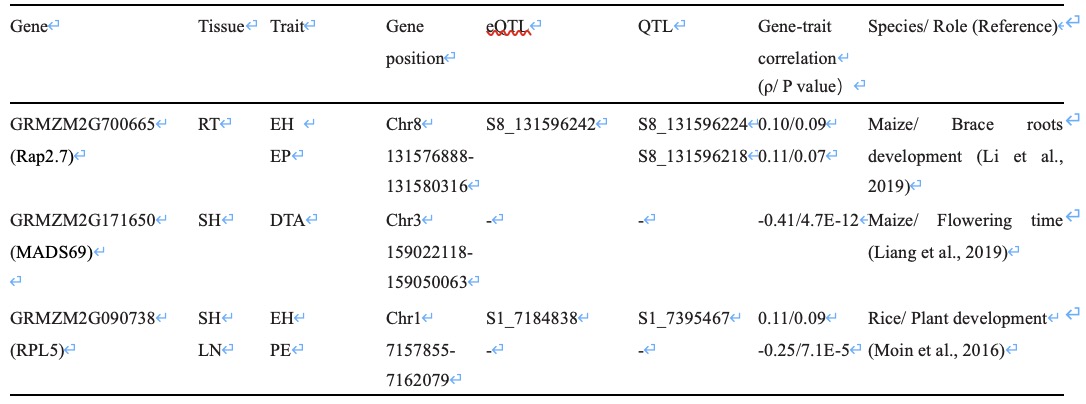
| Gene | Tissue | Trait | Gene position | eQTL | QTL | Gene-trait correlation(ρ/ P value) | Species/ Role (Reference) |
|---|---|---|---|---|---|---|---|
| GRMZM2G700665(Rap2.7) | RT | EH | Chr8 131576888-131580316 | S8_131596242 | S8_131596224 | 0.10/0.09 | Maize/ Brace roots development (Li et al., 2019) |
| GRMZM2G700665(Rap2.7) | RT | EP | Chr8 131576888-131580316 | S8_131596242 | S8_131596218 | 0.11/0.07 | Maize/ Brace roots development (Li et al., 2019) |
| GRMZM2G171650(MADS69) | SH | DTA | Chr3 159022118-159050063 | - | - | -0.41/4.7E-12 | Maize/ Flowering time (Liang et al., 2019) |
| GRMZM2G090738(RPL5) | SH | EH | Chr1 7157855-7162079 | S1_7184838 | S1_7395467 | 0.11/0.09 | Rice/ Plant development(Moin et al., 2016) |
| GRMZM2G090738(RPL5) | LN | PE | Chr1 7157855-7162079 | - | - | -0.25/7.1E-5 | Rice/ Plant development(Moin et al., 2016) |
All code needed to reproduce the results from this study are available on GitHub(https://github.com/leilei37/scripts).
References
LI J, CHEN F, LI Y, et al. 2019. ZmRAP2.7, an AP2 Transcription Factor, Is Involved in Maize Brace Roots Development. Front Plant Sci [J], 10: 820.
LIANG Y, LIU Q, WANG X, et al. 2019. ZmMADS69 functions as a flowering activator through the ZmRap2.7-ZCN8 regulatory module and contributes to maize flowering time adaptation. New Phytol [J], 221: 2335-2347.
MOIN M, BAKSHI A, SAHA A, et al. 2016. Rice Ribosomal Protein Large Subunit Genes and Their Spatio-temporal and Stress Regulation. Front Plant Sci [J], 7: 1284.