Bulked segregant analysis(BSA)分析学习分享
Bulked segregant analysis
综述 https://onlinelibrary.wiley.com/doi/10.1111/tpj.15646
所有方法都应考虑仅留取父母本本就存在的variant。
方法一
材料: X基因型株系、X基因型使用诱变剂产生的突变体(或随意亲本)
genome reference: X基因型
- 将材料杂交得到F1,F1自交得到F2,在F2中选择其中一种极端表型构建混池。
- 将混池和参考基因组进行比对并call snp得到vcf1文件。
- 此时vcf1文件中除了突变位点,其他部分都应与X基因生成的vcf2相同。
参考:https://academic.oup.com/g3journal/article/7/12/3947/6027434#235903430
可参考分析代码:https://github.com/qslin/Bulk-Segregation-Analysis/tree/master
材料:X基因型株系、X基因型使用诱变剂产生的突变体(符合隐形遗传),Y基因型(或随意亲本)
- Y基因型和参考基因组进行比对去除与X基因型相同的部分生成 Y.fa。
genome reference: Y基因型 - 将突变体和Y基因型杂交得到F1,F1自交得到F2,在F2中选择其中一种极端表型构建混池。
- 将混池和参考基因组进行比对并call snp得到vcf1文件。
原理:比对时,只有突变位点以及附近与突变位点连锁的部分全部比对不上,其他部分符合分离定律,X基因型片段与Y基因型片段应该均为50%。
- 此时vcf1文件中突变位点的snp-index=1,其他部分为0.5 与Mutmap分析相似
参考:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3664862/
文中图: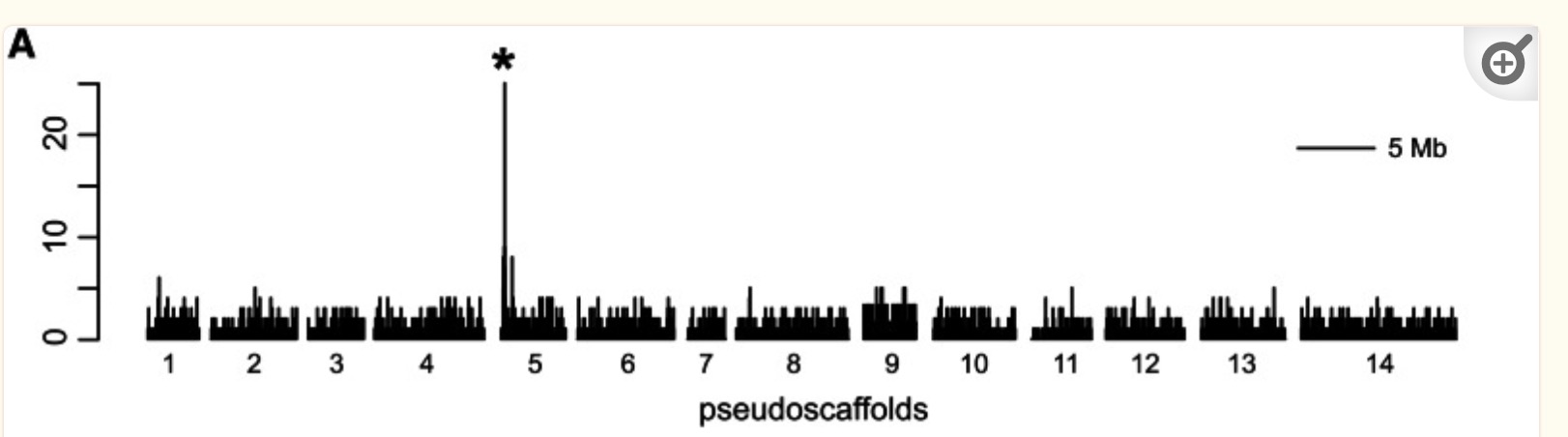
Genome scan for regions that are enriched in homozygous SNPs. Each pseudoscaffold of the M. lewisii SL9 (Y) genome was binned into 20-kb intervals, and the number of homozygous SNPs in each 20-kb interval was plotted in a bar graph.怎么理解homozygous SNPs?
方法二
Mutmap的Mutplot
与标准基因组进行比对,亲本之一和其中一个混池均得到bam进行后续分析得到delta SNP-index。
方法三
使用父本和母本杂交,得到F1,F1自交得到表型分离的F2。对F2建立混池,将两混池中SNP-index差异最大的染色体区作为候选基因区,使用母本或父本作为控制表型基因型的参考。
原理:https://onlinelibrary.wiley.com/doi/10.1111/tpj.12105
https://mp.weixin.qq.com/s/XFoSwo-HlPuu5sYKg82z8A 解读:
参照QTL-seq分析原理去理解,这里Reference sequence是亲本的基因组,不是call突变时用到的参考基因组序列。
比如1151这个位点,亲本是A,子代S-pool测到两种碱基,总深度是17,其中和亲本不一致的是C(频率是13),所以它的SNP-index就是13除以17;子代T-pool只测到一种碱基,总深度是10,只有一种和亲本不一致的C(频率是10),所以它的SNP-index就是10除以10,那么△SNP-index就是子代T-pool的SNP-index减去子代S-pool的SNP-index。同理去计算下面的。
SNP-index如何计算
以下研究结果例子来自QTL-seq test https://github.com/YuSugihara/QTL-seq
例1
vcf文件:
test_reference 118606 . T A 353.886 PASS DP=26;VDB=0.0891377;SGB=-3.69501;RPBZ=0.952352;MQBZ=-4.62481;MQSBZ=-0.72075;BQBZ=-0.411255;SCBZ=0;MQ0F=0;AC=4;AN=6;DP4=6,6,6,8;MQ=46 GT:PL:ADF:ADR:AD:GP:GQ 1/1:228,24,0:0,2:0,6:0,8:9.45607e-24,0.00613126,0.993869:22 0/1:50,0,213:3,1:5,1:8,2:3.874e-06,0.999996,3.23428e-22:54 0/1:113,0,118:3,3:1,1:4,4:1.9416e-12,1,1.02277e-12:115
0,2:0,6:0,8
3,1:5,1:8,2
3,3:1,1:4,4
snp_index.tsv
test_reference 118606 snp 10 8 0.675 0.5 0.8 0.5 -0.3
例2
test_reference 236239 . C A 190.98 PASS DP=35;VDB=0.650145;SGB=-2.03732;RPBZ=0.0391649;MQBZ=-5.28744;MQSBZ=0.539975;BQBZ=0.100739;SCBZ=0;MQ0F=0;AC=2;AN=6;DP4=12,13,5,4;MQ=48 GT:PL:ADF:ADR:AD:GP:GQ 0/0:0,39,255:6,0:7,0:13,0:0.999916,8.37425e-05,3.49839e-27:40 0/1:189,0,162:4,4:2,3:6,7:1.89242e-19,1,1.04935e-17:127 0/1:39,0,171:2,1:4,1:6,2:0.000189206,0.999811,1.32081e-18:37
6,0:7,0:13,0
4,4:2,3:6,7
2,1:4,1:6,2
test_reference 236239 snp 13 8 0.625 0.4904 0.5385 0.25 -0.2885
总结:
PL 归一化后各基因型的可能性,通常有三个数字用’,’隔开,顺序对应AA,AB,BB基因型,A代表REF,B代表ALT(也就是0/0, 0/1, and 1/1),由于是归一化之后,数值越小代表基因型越可靠;那么最小的数字对应的基因型判读为该样品的最可能的基因型;(provieds the likelihoods of the given genotypes)(具体计算流程?)
AD 两种碱基各自支持的碱基数量,用”,”分开两个数据,分别代表两个等位基因的深度。
参考:https://mp.weixin.qq.com/s/GmR7oeuPgpI2KhSYAOK1tw
重点:首先看parent PL项三个数字,如果第一个最小,就后续使用AD第一项/总AD;如果第三个最小,就后续使用AD第二项/总AD。
方法2,3步骤比较: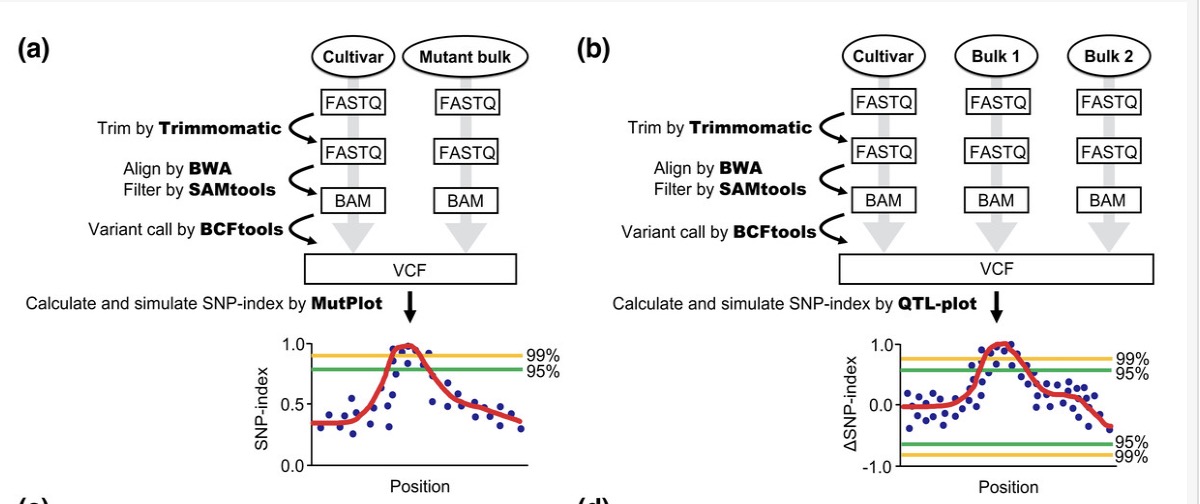
https://peerj.com/articles/13170/
note:call snp时,bam文件应该先合并再mpileup 还是直接mpileup?
https://www.biostars.org/p/237451/ 建议直接mpileup